The MDCG cybersecurity guidance – a helpful rush job
 It has been some time since the MDCG guidance on cybersecurity for medical devices was released (MDCG 2019-16 December 2019), so everybody has probably had the opportunity to get used to the document by now.
It has been some time since the MDCG guidance on cybersecurity for medical devices was released (MDCG 2019-16 December 2019), so everybody has probably had the opportunity to get used to the document by now.
While the document is by no means ideal or even flawless (congratulations MDCG on a glaring spelling mistake in the very title of the document (corrected later, just like the heading numbering mistakes)), it does provide helpful context on what the EU regulator would like to see in terms of cybersecurity for medical devices under the MDR and the IVDR, which concerns both standalone software and devices running software. The real value of the document is that it shows how different parts and requirements under the MDR and IVDR are interconnected via PMS and risk management, two areas in which the EU expects everyone to up their game considerably.
The document is based on IMDRF work on cybersecurity for medical devices, to which it often refers, specifically the very useful IMDRF’s Principles and Practices for Medical Device Cybersecurity document, which is currently under consultation.
The guidance comprises of
- An introduction;
- Basic cybersecurity concepts;
- Secure design and manufacture;
- Documentation and instructions for use;
- PMS and vigilance;
- Links to other EU and International legislation and guidance; and
- Annexes with examples and reference material
Some context
As you will know the MDR and IVDR contain new (cyber)security GSPRs in GSPR 17 and 16 respectively, but did you know that the MDR and IVDR require a much broader perspective on cybersecurity? As in many aspects, the MDR and IVDR do not provide a finite list of specific stuff you need to do, which, I know, is a problem for many manufacturers and especially their software developers.
A lot of software was never or is not developed from the ground up with cybersecurity in mind, and certainly not in the context of a good risk management plan that take into account all the risks related to its use in the environment that it is used in, the hardware it is run on, the dependencies it has with other systems and the relationship with other legislation that also govern software or IT processes, like the General Data Protection Regulation (GDPR).
Software developers scrum and sprint all over the place, often without having a good idea of the end that they should have in mind in terms of development goals. Often development teams will focus on delivering ‘something that works’ rather than ‘the software specified that meets the requirements specified’ and may even tack on some security at the end. Also, post market surveillance is typically not one of the things that is top of mind. Or, as researchers and authorities put it euphemistically (“MDMs” are medical devices manufacturers and MD is medical device):
“even if there is a clear regulatory framework for the introduction to the market of medical devices, the culture of cybersecurity is still very inconsistent among MDMs. MDMs fail to include cybersecurity in the MD design and development process as there is few guidelines or recommendations dedicated specifically to IT cybersecurity of medical devices.”
This approach will not do under the MDR and IVDR anymore. If there is one message that this guidance document gives, it’s that everything is connected (Connected Everything), that the MDR and IVDR require Consistency Everywhere (CE puns intended) and that the MDR acronym More Data Required also applies to cybersecurity, both pre- and post market.
What devices does this apply to? Well, to all devices governed under the MDR and IVDR, which includes standalone software that is a medical device under these two regulations, see MDCG 2019-11.
If you’d like to have an entertaining evening with cyber exploits of medical devices within a hospital, consider watching CSI Cyber’s Hack ER episode, in which a hacker takes control of all networked medical devices at a US hospital and threatens to kill one patient every hour if his demands are not met. Of course this is a fictional and much dramatized story, but the device and network exploits shown in it are all possible and do happen in reality. The ones in this episode are actually among the easier ones. This episode dates back to 2015 – hospitals are regularly hit by ransomware attacks and devices still get hacked all the time.
This may also be a good way to explain to your company’s management that lives are actually at stake if cyber risk management of the devices does not check out. Management is often less concerned with these technicalities and more focused on sales, as we’ve recently seen in the passenger aircraft industry: regulatory Cassandra and engineering Cassandra sound the alarm (this design is unsafe and does not meet requirements), regulatory Cassandra and engineering Cassandra are subsequently completely ignored or told to shut up, after which the risk does materialise and management bails with their golden parachute. Is that a responsible way to do things? As my grandmother would say: asking the question is answering it.
So let’s be responsible. Even if security by design is the best way to have safe technology, yet it is also an important concept under pressure because of the way technology is designed, developed and deployed. I have seen at many companies that design and development processes are just not set up or funded to deliver the best possible outcome in this regard. Under the MDR and IVDR this will not fly anymore, and substandard processes or outcomes will lead to non-conformities and/or enforcement.
An introduction
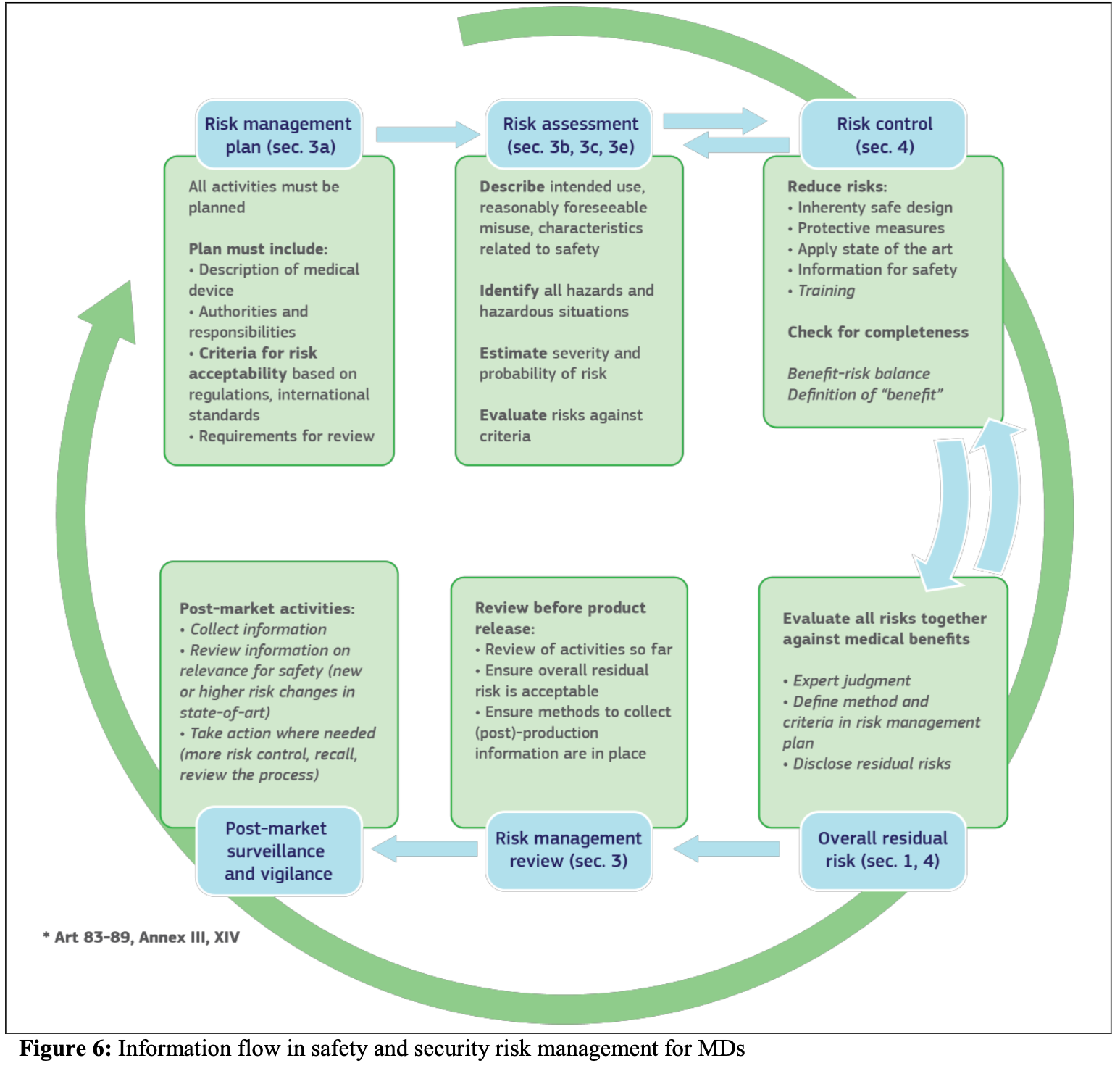
So how does cybersecurity function under the MDR and IVDR and how is everything interrelated? The below visual from the introductory chapter gives a nice overview.

This concerns a lot of processes, all of which are interrelated. I teach MDR these days by using the acronyms Connected Everything and Consistency Everywhere. This is definitely true for cybersecurity as well, because its requirements affect many processes and a lot of documentation horizontally and even diagonally:
- Conformity assessment procedures: Article 52 MDR / 48 IVDR
- Privacy and data protection: Article 62 (4) (h) MDR / 58 ((4) (h) IVDR: General requirements regarding clinical investigations conducted to demonstrate conformity of devices (this is not really addressed in the guidance but I’ve written something about it in this blog)
- Post-market surveillance system of the manufacturer: Article 83 MDR / 78 IVDR
- Post-market surveillance plan: Article 84 MDR / 79 IVDR
- Post-market surveillance report: Article 85 MDR / 80 IVDR
- Periodic safety update report: Article 86 MDR / 81 IVDR
- Reporting of serious incidents and field safety corrective actions: Article 87 MDR / 82 IVDR
- Trend reporting: Article 88 MDR / 83 IVDR
- Analysis of serious incidents and field safety corrective actions: Article 89 MDR / 84 IVDR
- Technical documentation: Annex II MDR and IVDR
- Technical documentation on post-market surveillance: Annex III MDR and IVDR
- Clinical evaluation and post-market follow-up: MDR and IVDR Chapter VI and Annex XIV MDR / XIII IVDR
Let’s take a look at risk management, because that is at the core of cybersecurity. Risk management is dealt with much more prescriptively in the MDR and IVDR than in the directive. GSPRs 1 to 8 in Annex I in both regulations hardwire risk management into the law much more prescriptively as happened under the directives, and there is a lot of risk management language scattered in the text of the regulations. You have to meet these requirements meet by default now (like the risk management system pursuant to article 10 (2) in both regulations).
The guidance ties the different parts of the MDR or IVDR that have to do with cybersecurity together, both visually and by means of a neat table, because everything is connected indeed.

The guidance also contains a convenient table for reference to Annex I related cybersecurity obligations. This is not the whole picture for the whole MDR and IVDR though, mind you, but it does show that the cybersecurity related measures are not only contained in sections 17 and 16 of Annex I of the MDR and IVDR respectively – and this table just shows the links within Annex I of the MDR and OVDR.

Basic cybersecurity concepts
Chapter 2 of the guidance discusses a number of important concepts in cybersecurity. The guidance revolves around the well-known CIA concept (Confidentiality, Integrity and Accessibility) throughout the lifetime of the device. These requirements are described quite well in the BSI White Paper on Cybersecurity for Medical Devices.
Compromised CIA might impact medical purposes as specified in the medical device definition in MDR Article 2.
The guidance defines 8 security practices, which largely overlap with the premarket considerations set out in the IMDRF’s Principles and Practices for Medical Device Cybersecurity document that is currently under consultation.
Operating environment and other stakeholders
The relationship with the operating environment is discussed in a number of places in this chapter of the guidance and elsewhere in it. The manufacturer must have a good understanding of the operating environment in which the device will be deployed to be able to implement appropriate layered defense measures independent of and not affected by the operating environment, to ensure that the medical device is designed and manufactured in a way that ensures that the risks associated with reasonably foreseeable environmental conditions are removed or minimized. This is why GSPR 17.4 MDR and GSPR 16.4 IVDR require that the manufacturer addresses what operating environment conditions the device has been designed for in order to work safely as intended. This also applies to modification of a device! The guidance refers explicitly to the IMDRF Practices and Principles document here, and section 3.6 of the guidance goes into quite a lot of detail on the operating environment.
One of the important and interesting parts is the interface between manufacturer and other stakeholders (systems integrator, operator, end user) emphasizing joint responsibilities. While I think it is very important that the MDCG stresses that cybersecurity is a collective effort, the MDCG shows in this section that it does not always completely understand how contracts between manufacturers and customers work.
The operator section (2.6.2) does not mention the concept of health institution defined in the MDR and IVDR, although it seems to refer to this concept because it describes the operator as the party procuring the device, which is strange. It would have made sense to link this, as this does happen in the IMDRF’s Principles and Practices for Medical Device Cybersecurity document.
Secure design and manufacture
Secure design and manufacturing begins with the end in mind, which means managing risks from the very start and keep managing risks throughout the life cycle. Did I say already that risk management under the MDR and IVDR has become much more detailed and prescriptive?
I find that many companies do not do a very good job at this. Security as a requirement is added to the process too late, or the development process is chaotic and badly documented and does not take security into account at all. Or, that happens too, nobody tells the software engineers about these requirements and they find out when the development cycle does not allow for sufficient time to get this right anymore.
Standards
One of the big problems with secure design and manufacture is that there are (stilll) no harmonised standards under the MDR available. This means that there are no standards that give an automatic presumption of conformity under the MDR and that manufacturers of MDR covered devices will need to do their own state of art assessment per GSPR, will need to include rationale in their technical documentation why the standard selected is the appropriate standard for the GSPR concerned and why the standard is state of art. Notified bodies will audit this rationale and will find non-conformity where the rationale is lacking.
COCIR has developed a convenient overview of relevant standards in its 2019 document Advancing Cybersecurity of Health and Digital Technologies:

The guidance contains a list of standards in Annex III of the guidance, which largely overlaps with the above COCIR overview but mentions a number of additional standards as well, such as IEC 82304-1 Health Software Part 1: General requirements for Product Safety and of course the good old EN ISO 62366 / ISO 60601-4 Usability Engineering.
Documentation and instructions for use
The guidance makes it very clear that the system for risk management required under article 10 of the MDR and IVDR, which is laid down in the documentation of Annexes I (GSPRs), II (technical documentation, contains the GSPRs) and III (technical documentation on PMS).
In addition there is the information supplied with the device, sections 23 (MDR) and 20 (IVDR) of Annex I. The guidance contains a detailed overview of the requirements. Mind you that EU MDR and IVDR risk management is very specific and strict on first applying risk management as far as possible via design and risk reduction and only then managing the acceptable residual risk by providing user information.
PMS and vigilance
One of the important, but in my view also stil underdeveloped items in the guidance, is PMS and vigilance with respect to cybersecurity. This is the area where I expect the market to have the steepest learning curve.
Under the MDR and IVDR PMS is much a more prescriptively active lifecycle process that must be laid down in the PMS plan based on article 84 MDR and 79 IVDR, which means that the manufacturer must continuously evaluate whether the cybersecurity measures applied for the device remain state of art. Just passively reacting to user complaints is clearly not the way to meet this standard – a manufacturer that can not demonstrate having a PMS plan in place with the elements in Annex III, 1.1 (b) MDR and IVDR will not be seen as compliant and his devices will not be seen as safe.
As the IMDRF puts it in the Principles and Practices document:
“As vulnerabilities change over time, pre-market controls designed and implemented may be inadequate to maintain an acceptable risk profile; therefore, a post-market approach is necessary in which multiple stakeholders play a role. This post-market approach includes various elements and include: the operation of the device in the intended environment, information sharing, coordinated vulnerability disclosure, vulnerability remediation, incident response, and legacy devices.”
For devices that run software and that are software the cybersecurity section will be a very important part of the PSUR or PMS reports (article 85 and 86 MDR, articles 80 and 81 IVDR).
Vigilance processes at the manufacturer will need to be set up in a way that they can recognize cybersecurity issues and can get to the bottom of the root cause, and can do this very quickly. This is why the IMDRF Practices and Principles document recommends setting up
“an incident response management policy and build an incident response team based on its product portfolio. The aim of incident response team is to provide appropriate capacity for assessing, responding to and learning from cybersecurity incident, and providing the necessary coordination, management, feedback and communication, for timely and pertinent action during the next incident.
Preparedness includes establishing an incident management policy, developing detailed incident response plans, building an incident response team, routinely testing and exercising incident response, and continuously improving this capability through lessons learned.”
Or in the words of the guidance: An effective and successful post-market cybersecurity surveillance program should include the following aspects (section 6):
- operation of the device in the intended environment;
- sharing and dissemination of cybersecurity information and knowledge of cybersecurity vulnerabilities and threats across multiple sectors;
- vulnerability remediation; and
- incident response
The guidance emphasizes the active elements of PMS that are underlined in Annex III too, stating that this must include handling and remediation of cybersecurity incidents and vulnerabilities reported through the post- market surveillance and vigilance systems shall be carried out conforming to the methodologies described in section 3.2 of the guidance, with regards to:
- Assess the need for reporting serious and non-serious incidents and of carrying-out field safety corrective actions;
- Enhancing security capabilities;
- Update the original Security Risk Assessment;
- Update the Verification and Validation;
- Update the original Security Benefit Risk Analysis; and
- Update the Technical Documentation.
Can you see how these requirements are life cycle commitments to make the device and its risk management better? Welcome to the MDR and IVDR because that is the expectation. You will never be done improving your device and its cybersecurity.
The guidance goes into some detail describing vigilance reporting formalities. As I will discuss in relation to the GDPR below these processes will need to be linked into the GDPR incident procedures, such as notably data breach procedures. Mind you that the GDPR can require super fast reporting and even reporting to data subjects (patients). The link between devices cybersecurity and GDPR data security is also very evident in the section about legacy devices of the IMDRF Principles and Practices document (section 6.6) which is an area discussed under GDPR guidance years ago.
Companies often tend to focus on a patch as a corrective measure, but just pushing out a patch may not be enough and other compensating controls may need to be deployed. Also, a patch may create other vulnerabilities, so even the patch itself and the patch delivery process is subject to risk management (e.g. can you trust your user to administer the patch in a timely fashion proportionate to the risk and what if they don’t?). The IMDRF Practices and Principles document has a helpful section on patches and patch processes in the overall discussion of vulnerability remediation in section 6.4.
Cybersecurity in clinical trials
While the focus in the guidance is very much on cybersecurity for devices as part of CE marking, the MDR and IVDR also have cybersecurity requirements for clinical and performance trials that you do not really hear anyone about. Interestingly this is also not discussed in the MDCG guidance in any level of detail, although this is an important subject. It also goes to the heart of your clinical trial agreements and CRO agreements.
Article 72 (4) MDR and 68 (4) IVDR contain requirements for security and confidentiality of personal data of trial subjects, or more precisely: appropriate technical and organisational measures shall be implemented to protect information and personal data processed against unauthorised or unlawful access, disclosure, dissemination, alteration, or destruction or accidental loss, in particular where the processing involves transmission over a network.
These measures are not subject to notified body conformity assessment but rather to member states competent authorities oversight and actually their inspection (article 72 (5) MDR and 68 (5) IVDR). They do not only apply to trials where an investigational device is tested, but also concern the set-up of any devices trial involving other non-investigational devices, e.g. home monitoring of trial subjects with wearables and apps.
Because cybersecurity in clinic trials is also much focused on confidentiality of patient data, this means that any process of the manufacturer must be closely intertwined with software and software device security processes. Since these processes are often facilitated through third parties (CROs), everyone has a role in this. Where the device itself is investigational, it should already be up to standards from a cybersecurity perspective, which should be checked as part of the approval process (article 71 (3) (c) and (e) MDR and article 67 (3) (c) and (e) IVDR: whether the measures planned for the safe installation, putting into service and maintenance of the investigational device are adequate and whether the device meets the requirements for a safe clinical investigation device (which includes solid risk management).
All of these measures must be made transparent: the application dossier under Annex XV MDR or Annex XIV IVDR contains (Annex XV, 4.5 MDR or Annex XIV,4.5 IVDR) a description of the arrangements to comply with the applicable rules on the protection and confidentiality of personal data, in particular (so not limited to or only):
- organisational and technical arrangements that will be implemented to avoid unauthorised access, disclosure, dissemination, alteration or loss of information and personal data processed;
- a description of measures that will be implemented to ensure confidentiality of records and personal data of subjects; and
- a description of measures that will be implemented in case of a data security breach in order to mitigate the possible adverse effects.
This means that also member states’ approval bodies need to be able to understand this.
Links to other EU and International legislation and guidance
The MDR and IVDR are not an island in how they prescribe cybersecurity requirements. The below overview of the Commission shows the whole picture, of which I will discuss some of the regulations and directives referred to.

GDPR
As we have seen above, the MDR and IVDR weave the GDPR in already where it comes to data and IT security in clinical trails, as well as for the investigational device design. But there is more.
The GDPR contains requirements regarding privacy by design and default (default being an interesting one in the light of Annex I 17.4 MDR and 16.4 IVDR about operating environment), which includes the security to ensure this, as you can see in the ENISA Handbook on Security of Personal Data Processing. For the English speakers, some ICO guidances on security under the GDPR can be found here, here and here. Mind you, the UK has left the EU, so the ICO has nothing more to say about the GDPR for the future so only past guidance is of any use. There is also more security related guidance under the GDPR at the Commission’s website.
The GDPR furthermore requires reporting and investigating data breaches in a time frame much shorter than normal vigilance reporting deadlines under the MDR and IVDR and the sanctions for not meeting this deadline can be quite severe. In severe cases, companies are also obliged to communicate the data breach to all data subjects involved without undue delay and under 72 hours.
A data breach for GDPR purposes and a serious incident for devices regulation will often overlap, which means that a company should have interfacing procedures to ensure that it does not act inconsistently. As you can see in the Annex II to the guidance (Examples of cybersecurity incidents/serious incidents), many of the cybersecurity incidents mentioned also involve a data breach or other form of access/processing that was not as intended. Therefore there should also be a very good connection between the company Data Protection Officer and the PRRC under the MDR and IVDR, to ensure that data protection related risk management and devices related risk management is consistent and adequate. Reporting requirements under the GDPR and MDR/IVDR also vary, which has to be accounted for in the procedures.
Keep in mind that where there are a number of parties involved (e.g. devices company, third party cloud provider, third party installation and maintenance services provider, health institutions and patients) in different jurisdictions in different time zones, things can get very complicated very quickly if you have not planned in advance and 72 hours is very very short then. Also, your customers may have additional reporting obligations, for example under the NIS Directive if they are EU healthcare institutions.
NIS Directive
Healthcare institutions are likely to be subject to the security obligations foreseen by the NIS Directive. As explained here, the NIS Directive had to be implemented by the means of national legislation by member States before May 2018.
In fact though, several member States have envisaged delays in its transposition and some especially with regard to the identification requirement of Operators of Essential Services, which normally would include healthcare institutions. While the situation appears promising, a further coordinative effort at a European level might be needed, in order to ensure a more coherent implementative approach.
See here for a good overview of NIS Directive obligations. While the devices industry is not subject directly to these, its customers (healthcare institutions) often will be.
The guidance contains a convenient Annex I with Mapping of IT security requirements to NIS Directive Cooperation Group measures.
Cybersecurity act
The EU Cybersecurity Act envisages the creation of a pan-European cybersecurity certification framework for ICT products and services. The healthcare sector may be perceived as one of the key sectors to be covered by one or more ICT security certification schemes. Nonetheless, significant steps need still to be taken to this regard.
An overarching European Cybersecurity Certification Group will be established and each country will need to appoint a certification supervisory authority. Creating these structures may cause additional fragmentation of, and/or overlaps with, existing security policies.
Whereas the cybersecurity certification in principle is voluntary, this can be overruled by any Union or Member State law. As a consequence, a patchwork of regulatory requirements may appear, as Member States introduce their own requirements for cybersecurity certification.
Overall
The document is a helpful piece of guidance for a more wholistic picture of cybersecurity requirements under the MDR and IVDR put in context. It feels however put together hastily, which is evidenced by the first published version containing formatting errors and typos even in the title of the document on the cover page. The document does not fit together as systematically as it could, which shows to me that also for the MDCG this is a developing item.
Although the MDCG guidance has helpful visuals in it, the IMDRF Practices and Principles document is set up much better and more systematically, and contains many useful and precise examples. Used together these documents are very useful.
Manufacturers that already implement very solid state of art software design may find that they need to up their game somewhat (this will not be the majority), but I am sure that there are many manufacturers that will need to make significant improvements, especially in the nexus with the GDPR and their PMS/vigilance procedures.
Health institutions would do well to understand the operator and end user dimensions of this, as knowledge and capacity is often lacking in my experience.
CROs and other service providers also need to understand these requirements.
This document should therefore be used in context with GDPR guidance, the other software guidance MDCG 2019-11 and the to be expected Guidance on clinical evaluation and performance evaluation of medical device software.