MDR and IVDR in 2019: up or out, sink or swim
 Happy new year and welcome to 2019, a truly decisive year for the medical devices industry with interests in the EU. If you haven’t spent any time so far getting ahead of events relating to the MDR and IVDR, this is the year that reality will start catching up with you. This year will determine if a manufacturer goes up, or potentially out.
Happy new year and welcome to 2019, a truly decisive year for the medical devices industry with interests in the EU. If you haven’t spent any time so far getting ahead of events relating to the MDR and IVDR, this is the year that reality will start catching up with you. This year will determine if a manufacturer goes up, or potentially out.
Why is 2019 so important? Read on, and you’ll agree with me that it is. First, take a look at the below diagram with the transition timelines, to which I have added my own embellishments (the red and orange rectangles):
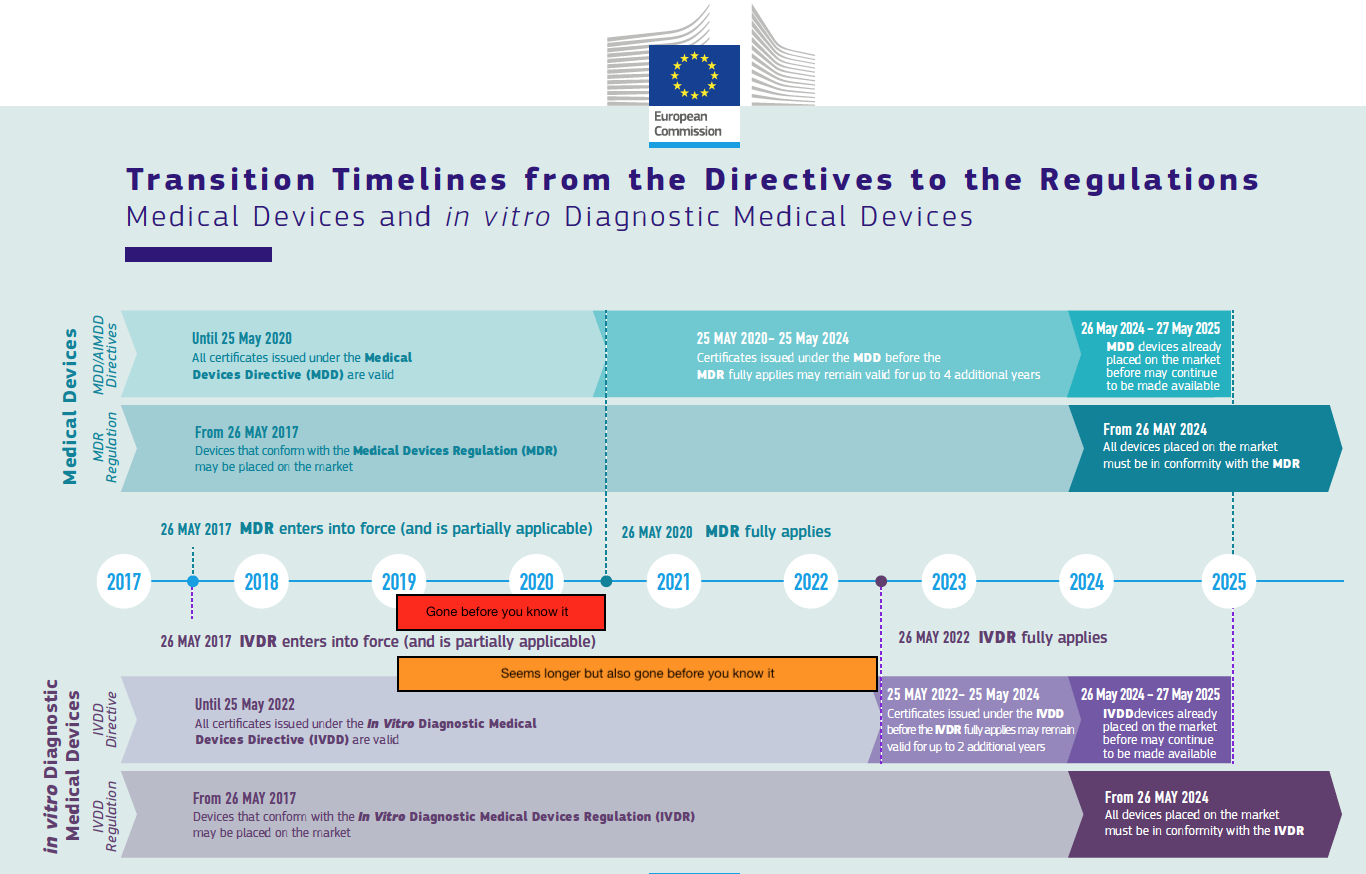
IVDs
IVD companies will be like: 2022 – so far away! Actually – no. 85% of the IVDs will need to be evaluated by notified bodies compared to 7% currently under the IVDD. This means that 85% needs to be IVDR certified by a notified body and that 78% of the IVDs on the market need to be CE certified by a third party for the first time. This will need to happen by notified bodies that have not been accredited yet (and will be much fewer in number than under the MDR), in accordance with new and stricter rules for performance evaluation. I bet that most of the technical documentation for currently self certified IVDs is a big leap away from where they need to be. IVD companies routinely forget that the IVDR impact on the IVD market is much much bigger than the MDR impact on the medical devices market and the notified body bottleneck much bigger. More on that in follow-up IVD specific posts. One thing at this point already: IVD companies should be well underway with their remediation in 2019, because the soft transition period is two years shorter, so in the end they must be fully compliant with the IVDR at the same time as the general medical devices must be compliant with the MDR.
“One Year of Application” – notified bodies’ perspective
Even the biggest optimists that like to shroud themselves in a cloud of mystery (the notified bodies) have started having open reservations about the regulatory system coming together. Team NB and NB-Med published a note “One Year of Application” in December 2018 that confirms the concerns I’ve been reporting about on this blog:
- Implementation period, May 2017 until May 2020, is too short for all stakeholders, taking into account that many details for both, manufacturers and Notified Bodies, are still under discussion
- Missing Guidance Documents enabling clear interpretation of specific requirements
- Unclear process behind the divergent opinions from the Joint Assessments
- Unharmonized interpretations of the Joint Assessment Teams and the various member states
- Capacity shortage for some medical device codes
- Workload for two legislative frameworks running in parallel for a period of time, from May 2020 until May 2024
In that note notified bodies state loudly and clearly that
- we have to count on not all products and manufacturers being certified under the MDR in time,
- capacity to renew (AI)MDD certificates for soft transition is limited (I read between the lines: ‘also possibly insufficient to renew all certificates timely’)
- it is not certain that all required class I R certificates (class I reusable instruments need a certificate under the MDR) will be granted in time because no notified body has been accredited for this yet.
Another interesting publication from Team-NB is the press release of 18 December “Surveys on MDR designation process steps”, which provides an update as to where the 22 Team-NB notified bodies that provided information are at present in the designation process:

The 22 Team-NB notified bodies are joined in the process (as per end November 2018) by 6 more non-Team-NB members that we know of, as per the amended joint assessment overview published by the Commission in December 2018. This overview states in relation to scope coverage “overall, the entirety of MDR and IVD codes”, which in my lawyer interpretation engine means: not everybody applied for full scope, and probably more than one did not. This makes it important for everybody to know if your notified body applied for the codes that you need.
This shows how (as I reported on this blog) incredibly over-optimistic to the point of being misleading the Team-NB notified bodies were around 26 November 2017, the date that the first applications for designation could be handed in – saying that all of them would immediately apply. It now mysteriously turns out that, for example, the Turkish notified bodies could not even ever apply for designation as Turkey has not transposed the MDR so far and two others have not even handed in their application. Yet, Team-NB was very sure at the time that all members would apply immediately on 26 November 2017, full scope. Scope is another question. While Team-NB says in the One Year of Application note that “All scopes, MDR and IVDR, are covered by the applications. Not a single product scope stays untouched.” I am not sure at all whether this can be interpreted as “every team NB member has applied for full scope MDR and IVDR and will be designated for it”. As “the expectations of the Joint Assessment Teams on resource qualification is dramatically increased compared to the requirements laid down by previous directives” it is by no means sure that notified bodies will come up short for designation (see One Year of Application note), or will have to drop specific codes from their application for which they do not have enough staff.
What is more: this data does not represent all Team-NB notified bodies and (by number) not the half of the notified bodies for the (AI)MDD (22 of 57 for MDD), which leaves big questions about the long tail of smaller notified bodies as the bigger ones are Team_NB members. Given that 6 more applied (and I’m assuming that there were none that applied for IVDR only) this means that 28 out of 57 MDD notified bodies applied so far. Less than half of the total number still. If you are with a notified body that has not applied by now, maybe think very hard about whether this one is right for you.
The notified bodies conundrum and resulting bottleneck is the biggest worry of industry, according to MedTech Europe.
Companies need to plan for two important notified body related windows in 2019:
- the date on which the notified body needs an application for recertification under the MDD or AIMDD for one last time, in order to benefit from the 2020-2024 regime; and
- the date on which the notified body needs to receive the MDR conformity assessment application, in order to be able to grant an MDR certificate before end May 2020.
For option 1, discuss with your notified body when this is. It looks like notified bodies will want to first do the MDD and AIMDD recertification applications (because of the insecurities in the MDR designation process), and subsequently the MDR work. BSI has indicated already it needs AIMDD and MDD recertification applications by end Q1 2019 (less than three months from now), but it may vary from one notified body to the other.
If you want an MDR certificate by May 2020 (option 2), count back 6-9 months (generally expected duration) from the date of application (26 May 2020) – this puts you between end August and end November 2019 under the most favourable circumstances. So one way or the other, you will be doing crucial stuff in 2019.
Did I already say 2019 was an important year? It will be an important year. You will remember it as the year in which the company made or broke its EU business.
Member states perspective
Member states keep generally taking the view that the system is ready enough but that they will keep monitoring especially notified body capacity coming online. An interesting account of a member state about the roll out of the EU system in a lot of detail (and with a lot of interesting sidebars to MDR and IVDR legislative history and EU level relevant quantitative information) can be found in the parliamentary documents for the Dutch MDR and IVDR implementation act, specifically the recent note after the first report (in Dutch, obviously – but easy to translate with Deep L, the excellent online translator that beats Google Translate hands down and offers privacy friendly options).
It’s interesting to learn for example that there is an explanatory ISO document for PMS under development and that the implant card format is still subject of inter-member state deliberation.
It also becomes clear now (as I’ve speculated before in presentations at conferences) that new MDR provisions that are not immediately regulatory still will be subject to competent authority oversight. An example is the requirement of sufficient coverage for product liability (article 10 (16) MDR and article 10 (15) IVDR), containing the mystic formula (see below image) So, the competent authority may ask a manufacturer about their coverage measures implemented and – in case it finds them insufficient – issue an administrative fine or penalty payment. It is my expectation that this will work no different in other member states. Since this applies as of the date of application of the respective regulation, better be ready by then and have some rationale about your application of the mystic formula set out in article 10 (16) MDR and 10 (15) MDR.

The Netherlands as a member state says it’s carefully monitoring the market for signals about possible shortages and disruptions in medical devices supply and how to deal with those, because the government is not sure if that has anything to do with the new regulations. The government says this development may also be caused by manufacturers changing strategy as a result of changing economic circumstances. This is one of those statements of which we say in Dutch (literal translation): “now breaks my wooden shoe”. I don’t think industry can be more vocal about the clear and present danger of this happening. [cue bullhorn sound filter:] “Hello Dutch government: the new regulations change the economic circumstances, and the manufacturers are adapting to that change. It’s really not that complicated, if you are just willing to drop your confirmation bias that the new rules do not cause massive changes. You have been sitting in at many meetings where this was expressed loudly and clearly. I see my clients shed loads of SKUs because it’s just not economically viable to transition them to the new rules. I see hospitals act surprised and be worried about this development. This shit is real, to put it bluntly. If you still do not get this, get over your bias – please.”
The Dutch government emphasises the importance of application of the General Data Protection Regulation on clinical data generated for the purposes of the MDR and IVDR, including incident reporting. Make sure to have your house in order in this regard, as I still see that a lot of manufacturers do not have a handle on this.
The rolling Rolling plan roll-out
As I’ve written in my previous post, a lot of roll-out of the CAMD Roadmap will happen this year. The roadmap did not have any real timing in it, but the rolling plan does, and the Commission has just mentioned that you should read them in conjunction.
It’s about time that the roll-out starts rolling, because there is quite some crucial stuff in there if you want to get your transition right. You might have been almost ready, but without the elements in the Rolling plan you cannot be fully ready, or be sure that you are. In that light it is pretty disappointing that the companies that gave it their all to be ready in time are not rewarded for their efforts.
Especially the Annex XVI non medical devices companies have a really raw deal – zero guidance so far, no common specifications yet (these are being developed and should become clear in the coming months). Just nothing to go on really. For them this transition period is truly not a transitional period at all.
If a company does not manage to roll with the rolling plan and is not ready to submit files to its notified body when it is designated or needs the application to manage workload, the company will likely miss being compliant by end May 2020. And that means market foreclosure after that date.
Brexit – still a very wild wildcard
As they say: “gouverner, c’est prévoir” – governing is looking ahead, precisely what the UK government is not doing with the Brexit dossier by delaying a vote on the Brexit deal negotiated with the EU but not yet accepted by the UK. Let’s sacrifice rationality to politics and populism just a little more, because who needs a functioning economy connected to its biggest international trading partners anyway, right? Even at the date of this blog all options are still on the table according to the papers: no deal Brexit, second referendum to reverse the Brexit, just accepting the negotiated result arrived at on 6 December. In other words, the whole spectrum. We are still nowhere, really.
In the storm of all these prudential politics the MHRA has amended its Brexit technical notice for medicines, devices and clinical trails on 3 January because while the MHRA remains committed to the EU internal market, it’s government certainly is not and a no deal scenario is still firmly on the table. According to the MHRA notice the most important and direct consequence on 29 March in a no deal scenario would be that none of the UK notified bodies would
“be able to assess the conformity of medical devices for devices to receive the CE mark and enter the EU market. Therefore, the MHRA will no longer be able to oversee Notified Bodies in the way that it does now.”
This is obscure and polite language for: all CE certificates issued by UK notified bodies will lose validity on 29 March in a no deal scenario. The UK may still allow these products on the market for some time, but no EU27 member state is obliged to do so (nor has said at this point affirmatively at this point that it will).
So, have you asked your UK notified body if it can switch over your certificates to its EU27 counterpart before end of March? If they cannot, or if you did not ask, all your certificates and the market access that they entitle your company to in the EU (also outside the EU, in countries that ask for a valid underlying CE mark for a national application) are at risk in case of a hard Brexit, so there is absolutely no cause for alarm.
Yes – in a no deal scenario, it’s back to the basics that the Commission outlined in its no-deal notice for goods already a almost year ago. Oh, you never read that, or the company never took this seriously? Well, the joke may be very much on you with less than three months to prepare for this realistic risk. After all, it’s only core business, right? You were maybe thinking that it’s very much like the annoying airplane briefing “in the unlikely event of a sudden loss of cabin pressure yadayadayada” (I think stewardesses may also feel like Regulatory Cassandra) – this will never happen so why even consider this as a scenario worth thinking about. Well there you might be on 29 March with the comfortable airplane having its door blown out and your company fumbling for the oxygen mask during explosive decompression of the airplane. Happy times!
So
Swim or sink this year, up or out for the MDR – however you want to see or call it. Companies that have not been taking preparation seriously so far will have an interesting year, in which they can make or break their EU business, and the outside EU business depending on the EU CE marks. That can be considerable too.
IVD companies should be underway as well, because the soft transition period is two years shorter, so in the end they must be fully compliant with the IVDR at the same time as the general medical devices must be compliant with the MDR (May 2024) while working with a bigger bulge to pass through the bottleneck of notified body capacity.
I’ll be here the whole year to help you weather the storm – ask your questions earlier than later though is my advice. Want me to yell at unresponsive management that doesn’t understand it’s only core business at risk? Let’s do that sooner than later too. Happy 2019!