Date of Entry into Force plus three years and a little bit

MDR DoA moved: industry be like
Last week we went past DoE (Date of Entry into force) of the MDR plus three years, and the date on which the MDR was supposed to have become applicable.
As you, my dear readers, will all know by now, the date of application (DoA) has been moved with a year now as the outcome of a thrilling rollercoaster that I described on this blog in much detail. It was kind of silent on the blog afterwards because from a legal perspective nothing much interesting has happenend since: the MDR was amended with the amending regulation and the DoA was moved up as projected. The consolidated version of the MDR with the changes incorporated is here. And I was very busy with all kinds of questions resulting from this move.
As I have blogged, the amending regulation does more than just move the DoA of the MDR, it also creates some unprecedented new powers for the Commission to take emergency approval measures under the existing directives and it even seemed to make it possible that Eudamed could be launched according to the agenda as planned.
So where does this leave us?
Industry response
Part of industry was quite happy with itself, because “it had won” – yay! Those with some formal education will be familiar with the concept of Pyrrhic victory, because that is what this is. It is merely delaying the inevitable. And those that have dealt with that know that the inevitable always happens.
Many other more responsible companies were very annoyed with the situation, as they had worked like crazy to be in time for the DoA, only to find that it had been moved up. For them this creates budget and resource problems, as staff needs to remain available to MDR projects for an additional year, which was not foreseen in the often hard fought MDR budgets and project timelines.
Rest assured that the DoA was not moved to give industry more time, it was moved to give notified bodies and competent authorities more time to complete the rollout and certificates conversion that they were much behind with. The enormous disruption of business as usual caused by COVID-19 created the political momentum for the change, because the Commission and member states realised that the epidemic would delay things so much it would be embarrassing and unworkable.
Quite some companies responded with the short term reaction that is characteristic for a three month horizon: many MDR projects were shelved and MDR transitions teams were disbanded or decimated because the ‘resources’ had been allocated to other projects and the company was not going to pivot on this. The DoA is a year away again, so why make the best of the extra time, right? In the end it’s only core business, so how could that be important. So many things you can do in one year, and so many loose ends to tie.
A lot of companies seem under the impression that the move of the DoA means that all the other subsequent deadlines at the end moved up with one year too. Wrong! The other deadlines of May 2024 and May 2025 did not move at all. So one year more until DoA is one year less of the four years of transition to convert the last MDD declarations of conformity and (AI) MDD certificates into MDR certificates and MDR Declarations of Conformity:
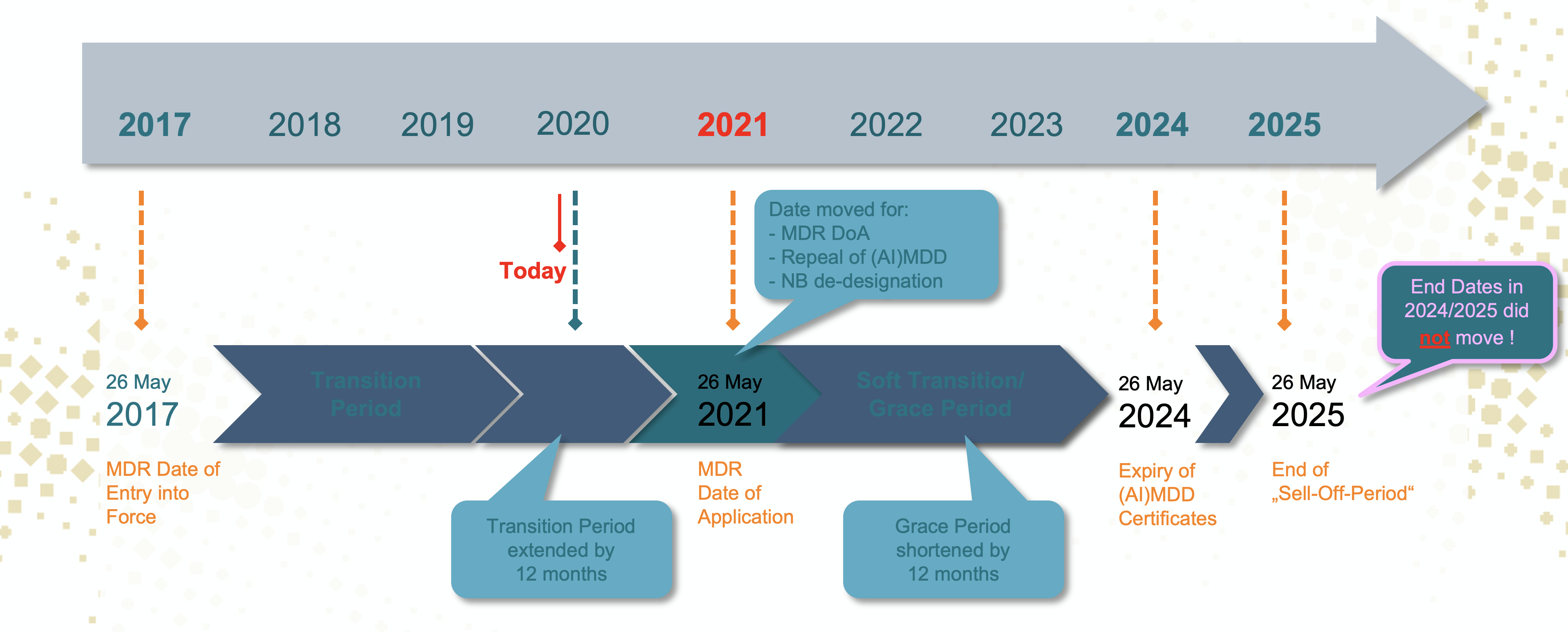
Does the delay help?
Yes and no, and in different ways than you might think. I invite you to watch Amanda Maxwell’s interview with Bassil Akra, Gert Bos and me for different perspectives on this matter, kindly made publicly available by MedTech Insight:
See for the interview https://medtech.pharmaintelligence.informa.com/MT142211/EU-MDR-Panel-Discussion-Why-An-Extra-Year-Is-Not-Really-A-Delay-For-The-MDR–How-To-Survive-Changing
In the extra year authorities are working on completing the outstanding agenda of implementing and delegated acts, Commons Specifications (Q4 2020 for Annex XVI products now and Q3 2020 for reprocessing of SUDs), MDCG guidances and, of course, Eudamed. The medical devices website will be transferred from DG GROW to DG SANTE, not a minute to soon since devices have been back with DG SANTE for a while now.
The notified bodies that are notified under the MDR will likely have a very limited add-on effect to the existing capacity, because they will be severely limited in what they can do. Apart from the ramp-up time to come to full capacity, they cannot travel or go to sites due to the COVID-19 epidemic, which means that they cannot complete new conformity assessment applications under the MDR. Yes: the new guidance on remote audits does not apply to MDR and it does not apply to new assessment applications in any event. Things will have to get worse before this gets better.
Notified bodies that were at risk of not completing pending re-certification under the (AI)MDD prior to DoA as to enable the manufacturer to benefit from the soft transition (and their customers) are the ones that seem to benefit the most from the move of the DoA. That is, if they had not planned for the DoA to not move. As you know, all existing notified body designations under the (AI)MDD would have become void by 25 May. With this deadline in sight, you can bet that notified bodies and their staff have done some planning of their own, resulting in staff having given notice already before the DoA move was even announced on 25 March 2020 and notified bodies having planned not to be designated by that date. The fact that notified bodies whose designation would otherwise have ended in the additional year can be conveniently renewed up to 26 May 2021 under a quickly adopted change of law, for which you find the guidance here (MDCG 2020-11), does not mean that these notified bodies can just scale up again if they had actually planned to close down or continue with a skeleton staff sufficient to do only surveillance of the (AI)MDD certificates during the soft transition period. The amended designation regulation also does not allow for designation of new notified bodies. The question remains therefore how much notified body capacity was really saved with the DoA move.
The notified bodies that already had their MDR designation (14 in total now) will also have had planning issues that makes it more difficult for them to stay at capacity (or increase this) for (AI)MDD. Also for them it was a reality that the (AI)MDD assessment business would switch to surveillance only by 26 May 2020 and they planned for that as well. New staff was trained only at MDR, and therefore only qualified for MDR. All the same, you say? Not how the law sees it. Just look at how many different classes of driver’s licenses or pilot licenses exist, even though it’s all sitting in a vehicle and steering in two or three dimensions. Different vehicles require different qualifications. Therefore, given everyone’s planning and the limited options created by the surprise additional of the extra year, your best option as manufacturer is to follow your original planning with the notified body and in cases where you would not have had your (AI)MDD certificates renewed in time, enjoy the possibility that the notified body may still be able to pull it off.
It helps the Commission and the MDCG
The delay does help the MDCG and the Commission. Important planning was immediately amended: no expert panels under the MDR in 2020 and more room to complete guidance. The first MDCG guidances are starting to appear in version 2 and the MDCG and Commission are cranking out guidances like there’s no tomorrow. Just in May and April this year nineteen (19!) guidance documents were issued on top of a bunch of other MDR related documents.
The delay of expert panels obviously does not help manufacturers that must follow the clinical evaluation consultation procedure. Article 120 (7) MDR literally says that as long as there are no expert panels, the procedure for new high risk devices subject to additional EU level scrutiny cannot be completed.
The delay also gives the Commission and the member states more time to clear the congested queue of MDR notified body designation. Remember: we have 44 applications, which so far resulted in only 14 designations. This means that there are still 30 (thirty!) notified bodies backed up in the application procedure. Obviously this is not completely the fault of the authorities, because many notified bodies did not apply at the first opportunity (26 November 2017) and quite a number took significant time to clear their CAPAs, but nonetheless: here we all are.
Standards
The extra year will not be enough to complete the standardisation mandate for the MDR / IVDR that has been put to CEN and Cenelec. Their decision whether they accept the standardisation mandate finally made officially on 15 May is foreseen now for 17 June, and even if CEN / Cenelec accept the mandate (which some parties have recommended that they actually don’t) it will most likely not be before the MDR DoA that the standards will be harmonised. Better plan to go into the MDR without harmonised standards.
The Article 59 powers
The amendment proposal allows for one interesting innovation and that is the applicability of article 59 MDR to the devices under the directives prior to the DoA. In other words, when a member state issues an emergency measure allowing a non-CE marked device on the market, the Commission may extend this measure to the whole EU on a mandatory basis by means of an implementing act, see below for my embedded presentation on this point:
[slideshare id=234839879&doc=vonlanthenwebinarerikvollebregt-200601192037]
Guidance for how the Commission will work with this exemption regime has also been published in the mean time, which confirms that this will really only be used in exceptional circumstances for indispensable devices, and is clearly not intended as a CE mark bypass or bottleneck management mechanism.
We will need to see now how the system works in practice.
Eudamed
As I have mentioned the amending regulation seems to make Eudamed phase-in as envisaged possible but only one year later. The Commission has taken the view that this is not what is going to happen: the fact that the deadline has been moved does not mean that its plans have changed. Also the most recent version of the Rolling Plan sets “2022” as Eudamed go live date.
So it will still be a phased ‘voluntary’ introduction of modules, with the actors module going first some months before the new MDR DoA (likely March 2021). We still don’t know whether the investment in the voluntary elements will not have to be re-done later at some point. However, it would be a very good opportunity to test company machine to machine interfaces and, more general, the company’s Eudamed procedures because you can start interaction with Eudamed likely as from March 2021.
The actor module gives the option for manufacturers, authorized representatives, importers as well as systems and procedure pack producers to enter their data in Eudamed and to acquire a Single Registration Number (SRN). The SRN is a very crucial element in Eudamed and under the MDR in general as you will need it for declarations of conformity and certificates. Since it is important that the SRN can be obtained before the date of application of the MDR, the actor module will be first and before the new DoA. May 2021 is expected to bring two more modules: the UDI/Device Registration module and the Certificate/Notified Body module, which are also required for data entry and for data on certificates. The rest of the modules (clinical investigations, incident reporting and market surveillance) will follow later as these are intended as tools for communication about devices and actors. As envisaged under article 123 (3) MDR existing systems will be used for this until Eudamed is fully functional.
IVDs: a crisis in the making
I’ve said it before and will keep repeating it: the IVDR is a crisis in the making. Several developments conspire to make this a really big crisis that may put a serious dent in healthcare. Maybe some people noticed that in these epidemic times it is pretty sweet and kind of essential to have in vitro diagnostic tests that are actually validated. So why miss all these opportunities to plan ahead?
We had some guidance about COVID-19 test development under the current IVDD, which was a nice reiteration of what every serious IVD company that takes performance evaluation seriously should know already. If you’re an IVD company, it might be good to put your existing technical documentation along that ruler because this is the bare minimum that a notified body will expect under IVDD standards (as you know the IVDR will be a step up, with additional clinical performance data required).
At the moment there are de facto only two notified bodies designated for the IVDR (three in NANDO at the moment, but BSI has one in the Netherlands and one in the UK, the latter of which will lose its designation end of this year as a result of Brexit). It is not exactly clear how much of the required capacity these two notified bodies represent but given that there are still 12 notified bodies in the pipeline according to official figures, this is probably not as much as needed (the capacity that applied represents 62% out of the original number of NBs for the IVDD says the Commission, counting one extra new applicant, which is likely NSF that also abandoned its application again).
If you count back from the DoA of the IVDR, manufacturers must have an IVDR conformity assessment application in the door at a notified body by the end of 2020 in order to have a good chance of an IVDR CE certificate before IVDR DoA. As I have explained on previous occasions, the IVDR is different from the MDR in that most manufacturers on the market do not already have a CE certificate under the IVDD, while the large majority of them will need that under the IVDR and they have two notified bodies to choose from. This means that if you’re an IVD manufacturer you must plan for an IVDR conformity assessment application before the end this year. Let’s just hope more notified body capacity becomes available, but I am not optimistic. This means that when the two current IVDR NBs are full, the rest is largely out of luck. Yet, the sense of urgency seems to be missing, both on the side of industry in general and on the side of the authorities.
The IVDR has been structurally left out of any COVID-19 motivated emergency measures, such as the extension of article 59 MDR to the directives. It would have been a small matter legally to have included article 54 IVDR in this as well, thus allowing for emergency possibilities for IVDs. This would be extra important in the light of the bottleneck for IVDs going into the IVDR. Yet, apparently all eyes appear to be on the MDR at the moment.
So far no substantial guidance has appeared for the IVDR and half of the IVDR guidance on the planning is not even scheduled for 2020. This is why I am personally very happy with the new MedTech Europe guidance document “Clinical Evidence Requirements for CE certification under the in vitro Diagnostic Regulation in the European Union”, This document is a collection of questions and answers designed to help manufacturers navigate their performance evaluation obligations under the IVDR.
So what to do?
If I were a devices manufacturer I would not treat this extra year as an extra year of MDR DoA delay because it really isn’t. At best you have some months extra to fine tune and that may be welcome. Your MDR specialist might take some well-deserved holidays, but keep your eyes on the ball and don’t forget that everything moves slower in an ongoing pandemic so what would normally get done in the same amount of time will now take longer.
If I were an IVD manufacturer I would go full pull to have my technical documentation for my IVDs ready for the IVDR on the triple double and make sure to have my first conformity assessment applications in the door at a notified body this autumn to be ahead of the big congestion that is coming. Same is true here: expect things to move slower than normal because of the pandemic. We may even get a second wave that’s worse. Have you planned for that? Scenario anyone?